
Grupo de investigación dirigido por Xosé Bustelo, del Centro de Investigación del Cáncer (centro mixto de la Universidad de Salamanca y del CSIC)
Descubierto en Salamanca un nuevo gen inductor de cáncer y sus debilidades terapéuticas
La investigación ha sido desarrollada por el grupo de investigación dirigido por Xosé Bustelo, del Centro de Investigación del Cáncer (centro mixto de la Universidad de Salamanca y del CSIC)
16 marzo, 2022 12:32Noticias relacionadas
La secuenciación intensiva del genoma de tumores realizado a lo largo de estos últimos años ha permitido identificar miles de alteraciones genéticas, denominadas mutaciones, presentes en los mismos. Hoy en día, el gran reto científico es determinar cuáles de estas mutaciones son relevantes en el desarrollo del cáncer y, tras ello, descubrir los cambios que provocan en las células normales de nuestro organismo para convertirlas en células malignas. Dado los miles de mutaciones identificadas, este trabajo se puede considerar no ya análogo a encontrar una aguja en un pajar si no, más bien, a encontrar las agujas correctas entre de miles de agujas diferentes. Para complicar más este proceso, la mayoría de alteraciones genéticas presentes en los cánceres humanos se encuentra a muy baja frecuencia, lo que dificulta enormemente establecer su relevancia usando exclusivamente pacientes humanos. Abordar estos retos es importante no solo para entender la causa del cáncer, sino también para la implementación efectiva de la medicina personalizada, la cual se basa en el diseño de terapias en función del patrón de mutaciones que exhiben los tumores en pacientes específicos.
En el trabajo publicado hoy en la prestigiosa revista Cell Reports, y desarrollado por el grupo de investigación dirigido por Xosé Bustelo, del Centro de Investigación del Cáncer (centro mixto de la Universidad de Salamanca y del CSIC), ha podido demostrar que una mutación del gen RRAS2 identificada en los estudios genómicos de tumores humanos actúa como un inductor del cáncer en un amplio espectro de células de nuestro organismo. A través del estudio de las células tumorales generadas tras la expresión de esta versión mutante, este trabajo ha iluminado también los cambios que provoca en cada uno de los tipos celulares que originan dichos tumores. Esto ha permitido descubrir talones de Aquiles en cada uno de estos tumores lo que, a su vez, ha llevado a identificar fármacos que podrían ser usados para tratar a pacientes con tumores que alberguen mutaciones en dicho gen.
El cambio de comportamiento crítico de una molécula
La mutación que ha sido estudiada por estos investigadores representa una alteración muy pequeña, ya que implica el cambio de una única letra (nucleótido) de las 82.000 de las que está compuesto el gen RRAS2. Sin embargo, este pequeño cambio es crítico, puesto que hace que la molécula codificada por este gen cambie su comportamiento de forma radical. Mientras que la versión normal de esta molécula funciona como un interruptor que puede encenderse o apagarse dependiendo de la presencia de diversos mensajes extracelulares, la versión mutante está permanente anclada en el estado activado, lo que la hace funcionar de forma crónica sin poderse apagar nunca. Ello hace que estas moléculas mutantes manden señales de forma ininterrumpida, lo que provoca la proliferación descontrolada de las células que albergan mutaciones en este gen. Esta división continuada es lo que provoca, con el tiempo, la formación tumores en distintas partes del organismo.
La selección de esta mutación para la realización de este estudio se basó en varias pistas: en primer lugar, el hecho de que se detectaba de forma recurrente en tumores humanos, aunque a baja frecuencia. En segundo lugar, a que estudios previos del laboratorio habían mostrado que esta mutación generaba moléculas de RRAS2 con una activación crónica; en tercer lugar, porque se habían descubierto mutaciones en este gen en una enfermedad rara conocida por síndrome de Noonan, lo que sugería que las mutaciones en este gen debían tener una función patológica determinada en vez de surgir simplemente por azar. Un impedimento importante para su estudio, sin embargo, era que la baja frecuencia de estas alteraciones genéticas hacía imposible realizar estudios de causalidad usando grupos de pacientes de cáncer.
Para establecer el papel real de esta alteración genética en procesos tumorales, el grupo de Xosé Bustelo decidió generar un ratón modificado genéticamente en el que se esta mutación se podía inducir a voluntad de los investigadores en estadios postnatales. Con esta estrategia, se quería mimetizar lo más exactamente posible lo que ocurre en los estadios más tempranos de los tumores humanos: la aparición de una alteración genética determinada en las células sanas de un órgano adulto de un individuo y, a partir de ahí, ver cómo estas células evolucionan a largo plazo.
“Esto nos permitió descubrir que la inducción de esta mutación promovía la aparición de tumores en distintos tipos celulares y órganos como los linfocitos, el ovario, el testículo o la piel. El desarrollo de estos tumores es muy rápido, lo que nos indica que los tumores generados tras la inducción de la mutación en el gen RRAS2 no necesitan probablemente ninguna otra alteración genética para poder desarrollarse de forma efectiva”, indica Isabel Fernández-Pisonero, autora principal de este trabajo.
“El análisis posterior de las células tumorales procedentes de cada uno de estos cánceres nos permitió conocer los cambios que la mutación en RRAS2 inducía en el comportamiento de las células originaban los tumores y, como consecuencia, descubrir sus vulnerabilidades terapéuticas”, indica Xosé Bustelo, el investigador responsable de la coordinación de este estudio. “Estas investigaciones nos permitieron también identificar qué dianas y fármacos serían los más adecuados para eliminar los tumores con mutaciones en este gen. En concreto, hemos visto que la gran mayoría de los tumores inducidos por RRAS2 tienen como talón de Aquiles principal una molécula denominada mTORC1 para la cual ya existen fármacos disponibles. De acuerdo con ello, nuestro trabajo ha demostrado que la administración de inhibidores contra mTORC1 permite eliminar de forma efectiva la gran mayoría de los tumores inducidos tras la expresión de la forma mutante de RRAS2 en ratones”, añade Laura Clavaín, otra de los investigadores que han participado en este estudio.
RRAS2 es muy similar estructuralmente a una familia de genes, denominados como RAS, los cuales están muy frecuentemente alterados en tumores humanos. De hecho, uno de estos genes fue el primer gen inductor de cáncer descubierto en humanos gracias al trabajo del laboratorio de Mariano Barbacid en la década de los 80 del siglo pasado. Pese a dicha similitud, los resultados obtenidos en el trabajo publicado en Cell Reports indican que la mutación de RRAS2 induce un espectro de tumores diferente a los generados por mutaciones similares en los genes RAS. Las vulnerabilidades terapéuticas de estos tumores son también diferentes. “Esto nos dice que estas moléculas implicadas en cáncer se comportan más como primas lejanas que como hermanas gemelas”, indica Fernández-Pisonero.
Gracias a este trabajo, por tanto, se ha podido demostrar que las mutaciones en el gen RRAS2 sí son relevantes cuando se detectan en tumores y, además, se ha establecido unas posibles pautas para poder tratarlos con fármacos antitumorales ya disponibles en el mercado. “Eso sí, ahora queda demostrar que estos datos son extrapolables al ámbito clínico, para lo cual se tendrá que reclutar un número amplio de pacientes. Esto es importante porque el efecto de las terapias que se han visto efectivas en nuestro modelo animal puede alterarse por la presencia de las múltiples mutaciones en otros genes que acostumbran a encontrarse en los tumores humanos”, añade Xosé Bustelo.
De utilidad también de cara al síndrome de Noonan
Fuera del ámbito del cáncer, los resultados del presente trabajo pueden ser también de utilidad para saber los efectos que las mutaciones del gen RRAS2 inducen a nivel embrionario para generar la enfermedad congénita conocida por síndrome de Noonan. Esta enfermedad, que se origina tras el desarrollo de mutaciones en genes específicos en las primeras fases del desarrollo embrionario, está asociada a problemas en el desarrollo de la cabeza y de los sistemas circulatorio, muscular y nervioso que, en última instancia, afectan muy severamente a la calidad de vida y supervivencia a largo plazo de los individuos que la padecen.
“Nuestro trabajo indica que los individuos afectados de este síndrome probablemente tengan una alta tendencia a desarrollar algunos tipos tumorales a medida que avanzan en su edad adulta. También ha revelado qué tipo de fármacos podrían ser de interés para corregir algunos de los problemas médicos que manifiestan los individuos que padecen este síndrome”, indica Fernández-Pisonero. Aunque este estudio necesitará trabajos adicionales en el futuro próximo, “el modelo animal desarrollado en el presente trabajo nos da una ventana única con la que estudiar cómo se desarrolla el síndrome de Noonan a lo largo del periodo embrionario y qué oportunidades este proceso nos ofrece para desarrollar terapias que palíen al menos algunos de los problemas clínicos más graves de las personas que padecen este síndrome”, añade Xosé Bustelo.
El trabajo publicado ha sido el resultado del trabajo del grupo de investigación liderado por Xosé Bustelo (Centro de Investigación del Cáncer de Salamanca, Centro de Investigación Biomédica en Red de Cáncer y Conexión-Cáncer del CSIC) en donde han colaborado también los grupos de Dolores Caballero (Centro de Investigación del Cáncer de Salamanca, CIBERONC y Hospital Universitario de Salamanca) y Balbino Alarcón (Centro de Biología Molecular Severo Ochoa, CSIC, Madrid). El trabajo ha sido posible gracias a la financiación de la Asociación Española Contra el Cáncer, la cual ha apoyado el proyecto cooperativo a lo largo de los últimos cinco años y los contratos de varios de los componentes del grupo de Xosé Bustelo que han participado en este trabajo (Laura Clavaín, Javier Robles-Valero). Otras fuentes de financiación incluyen ayudas de la Junta de Castilla y León, la Agencia Estatal de Investigación y la Fundación La Caixa.
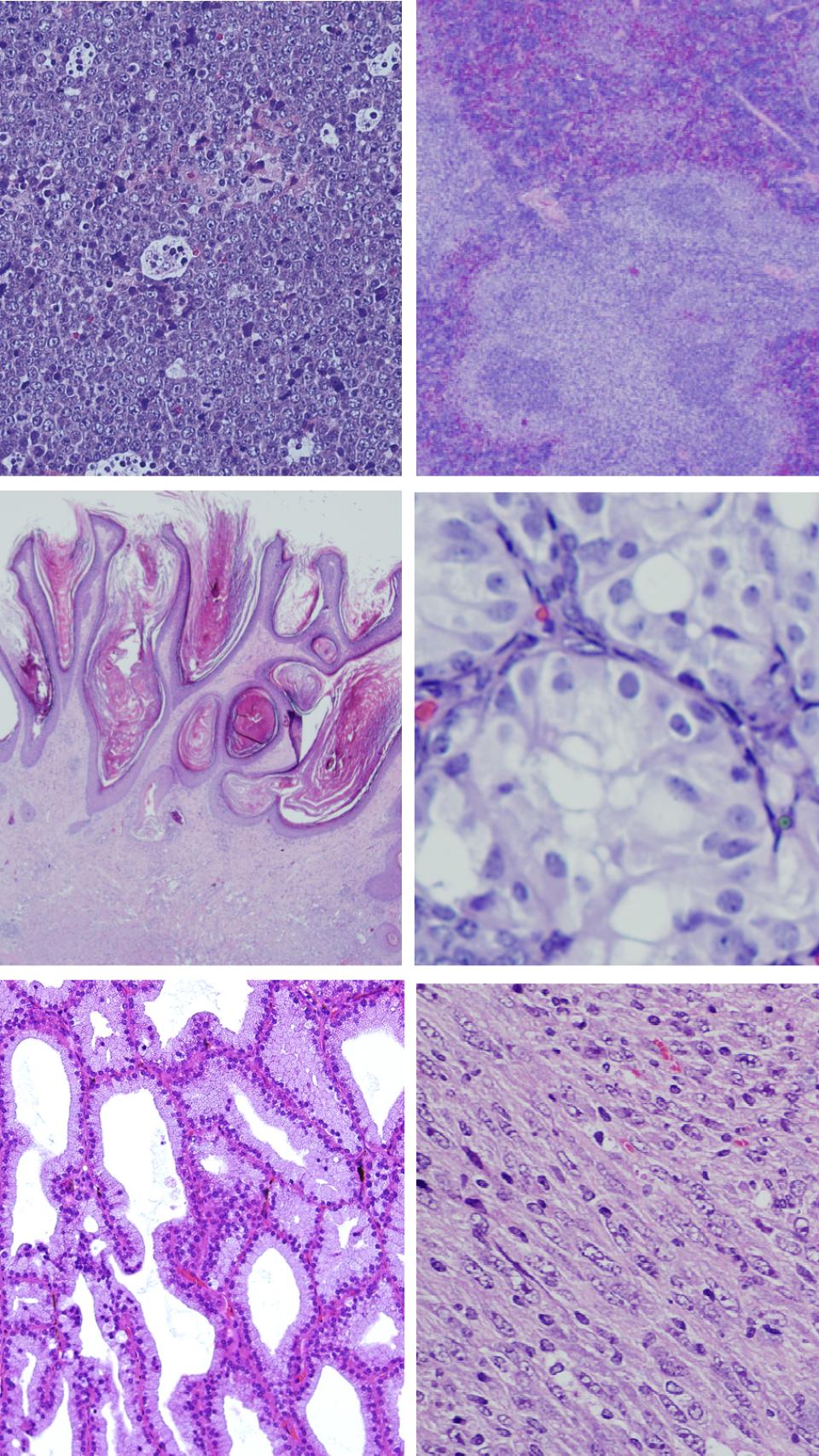
Ejemplos de los tumores inducidos tras la expresión de la versión mutante de RRAS2





